|
Current Level |
||||||||||
|
|
||||||||||
|
Previous Level |
||||||||||
|
|
||||||||||
|
BIO 460 Bioinformatics Protein Structure/Prediction General Properties of Amino Acids Classification of Amino Acids Levels of Protein Structure Protein Folding Prediction of Secondary Structure (PELE) Prediction of Motifs/Domains (PROSITE) Hydropathy Index (GREASE) Prediction of Transmembrane helices (TMHMM)
General structure Amino Acid Structure
Three central atoms
Ampholyte polyprotic acid with both basic and acidic groups
Zwitterion dipolar molecules at physiological pH
L-configuration
Classification of Amino Acids Non-polar amino acids
Polar
Charged Basic positively charged
Acidic negatively charged
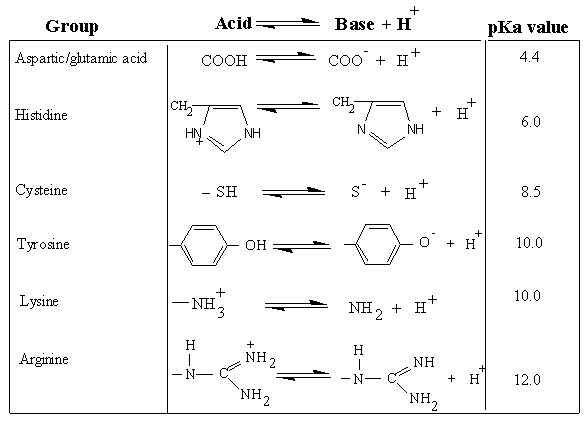
Acid/Base Properties of Amino acids
Ampholytes acidic and basic groups
Ionization of Various R-groups
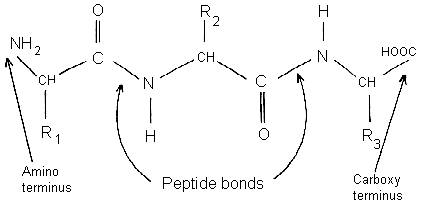
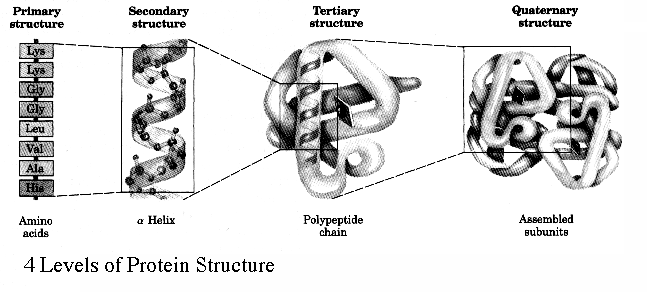
Levels of Protein Structure Primary structure the amino acid sequence of its polypeptide chain
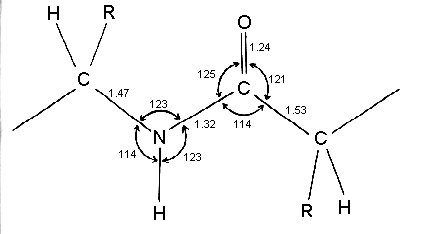
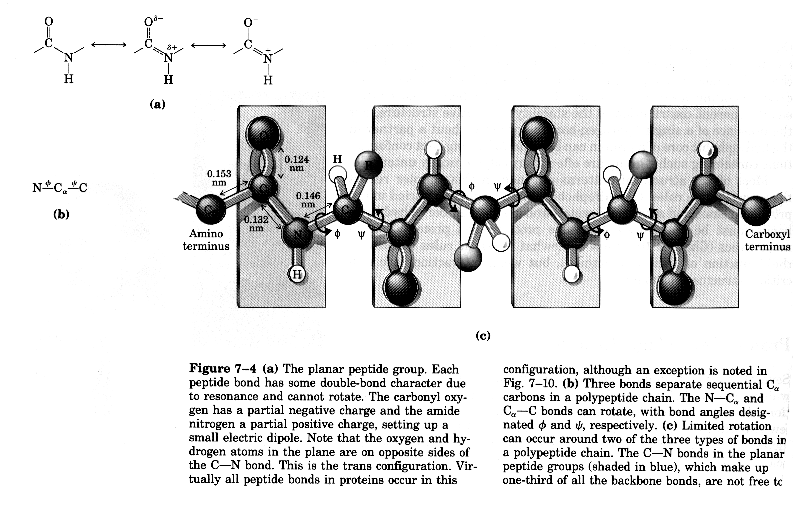
Types of bonds - Covalent peptide bond
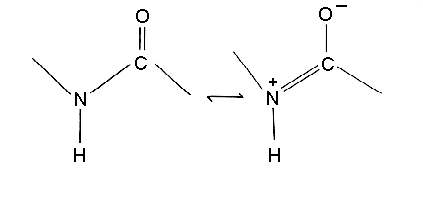
Peptide bond is rigid due to resonance
Geometry of peptide bond
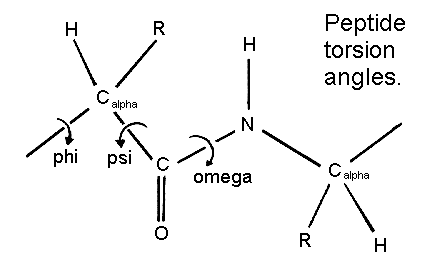
Another example of peptide backbone and its arrangement of atoms Torsion angles
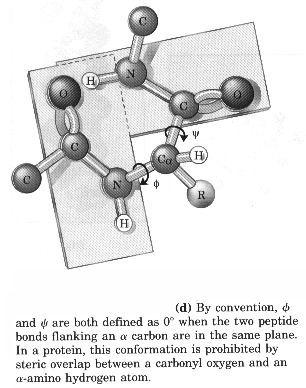
Starting convention for phi, psi angles
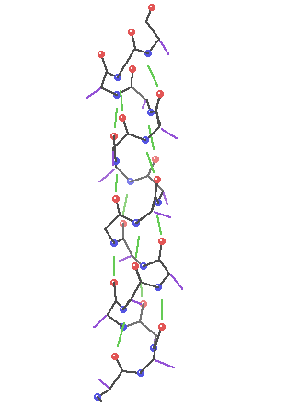
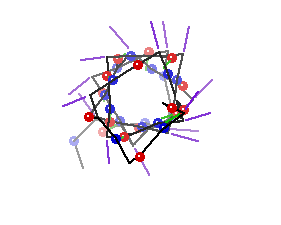
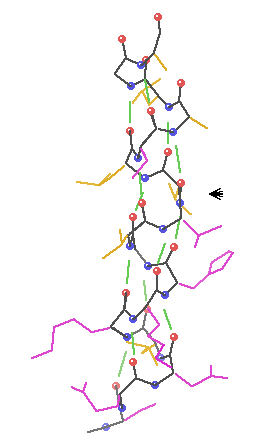
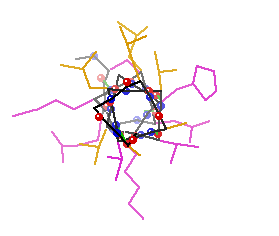
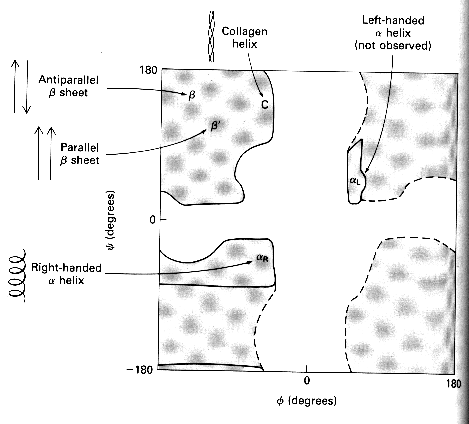
Secondary structure involves regular patterns of polypeptide folding including helices, sheets and turns torsion angles describe the possible regular conformations between adjacently linked amino acids there are only a few select sets of f and y angles that may exist between adjacent amino acids these angles are limited by both the rigidity of the peptide bond and the physical bulk of the R-group a-helix right-handed helical conformation has 3.6 amino acids per turn Properties of a-helix 5.4 Å rise per turn, 3.6 amino acids per turn, n, (n+4) hydrogen bonding pattern, 1.5 Å rise per amino acid
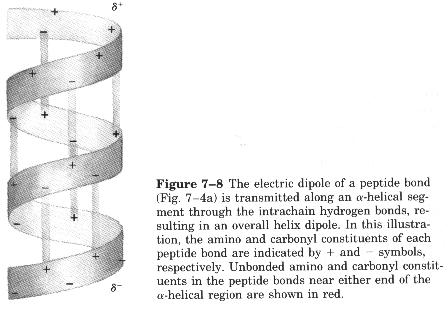
Characteristics of a-helices Right-handed 3.6 aa/turn 1.54 Å rise/aa 5.4 Å rise/turn R-groups point out away from the center of the helix Backbone hydrogen bonds are arranged peptide C=O bond of the nth residue points toward the N-H group of the (n + 4) residue Dipole moment within the helical axis (amino end more +) (carboxy end more -)
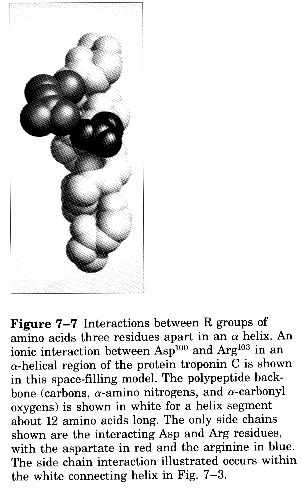
R-groups can form ion pairs along the helix
Dipole moment set up by the periodicity of the helix (amino end slightly positive, carboxy end slightly negative)
Amphipathic Nature one side of helix is lined with polar/charged sidechains the other with nonpolar (in the top view to the right the non-polar sidechains are lining the top side the polar/charged sidechains the bottom side)
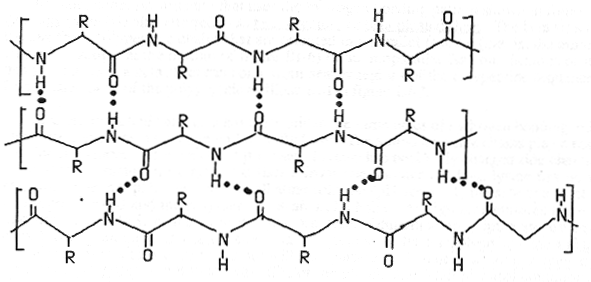
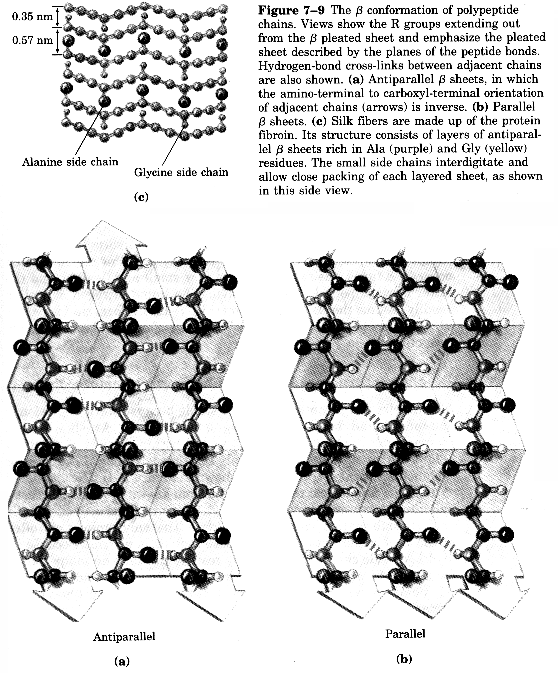
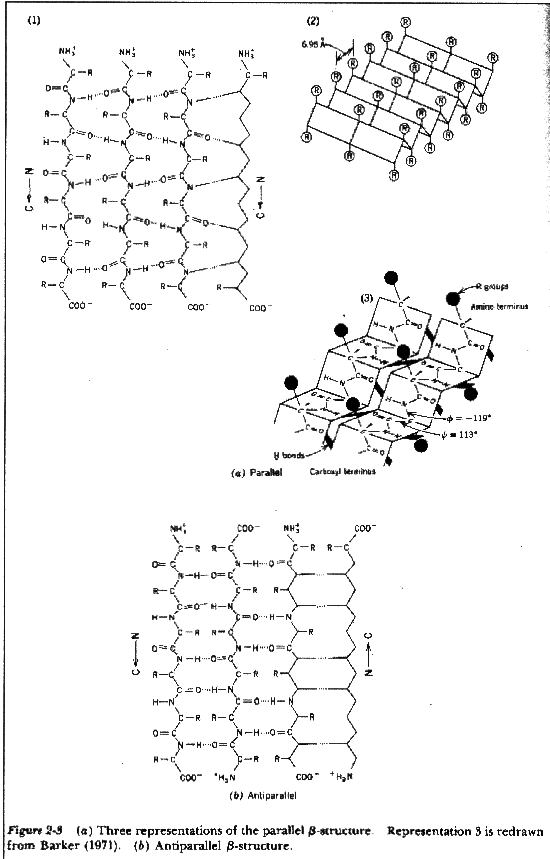
b-sheets hydrogen bonding occurs between adjacent polypeptide chains inside of within the same chain as in a-helices
Two Types of Sheets Antiparallel neighboring hydrogen bonded polypeptide chains run in opposite directions Parallel neighboring hydrogen bonded chains extend in the same direction
Characteristics of b-sheets Hydrogen bonds shared between neighboring chains Successive R-groups extend to opposite sides of the sheet and are located 7.0 Å apart Exhibit a right-handed twist Parallel sheets are less stable than anti-parallel sheets Topology (connection) between neighboring anti-parallel can be small and simple Topology between neighboring parallel strands is typically long and complex
Other views of sheets
Still more way to represent b-sheets
Turns and Loops used to join helices and sheets together allow protein structure to quickly change direction and keep its compact shape b-turns(bends) involved when peptide chain rapidly reverses direction see figure below Characteristics of b-turns Tight 180 ° turn 4 amino acids involved Glycine and proline are usually involved Glycine because it is flexible Proline because it can adopt a cis configuration Glycine usually residue 3 Proline residue 2
Omega (W )Loops have a necked-in shape of the greek uppercase letter omega (W ) Characteristics of omega loops
Compact R-groups tend to fill their cavities Located at surface of protein Involved in biological recognition
Ramanchandran Plot indicates allowed conformations of proteins most of the areas within the plot below (white regions) are not observed within protein structures exceptions are at place in the protein where there are Pro of Gly
Probability of certain amino acids in the various types of secondary structure
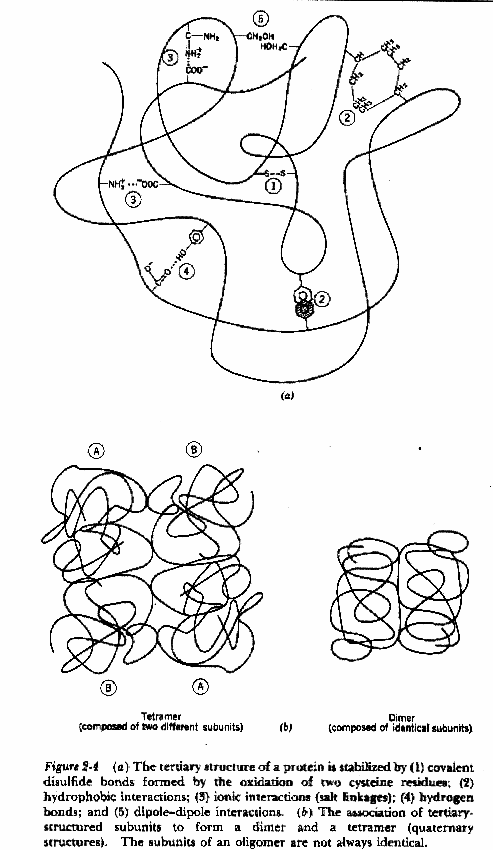
Tertiary structure describes the folding of the secondary structural elements and specifies the position of each atom in the protein these structures have come into view using either X-ray crystallography or Nuclear Magnetic Resonance (NMR) the coordinates for these proteins are located at the Protein Data Bank (PDB) and can be accessed via the web at (http://www.rcsb.org/pdb/) Types of bonds long and short range, covalent and non-covalent Covalent Disulfide covalent bond shared between twoadjacent cysteine sidechains Noncovalent Hydrogen bonds Hydrophobic interactions Van der Waal's interactions Ionic interactions
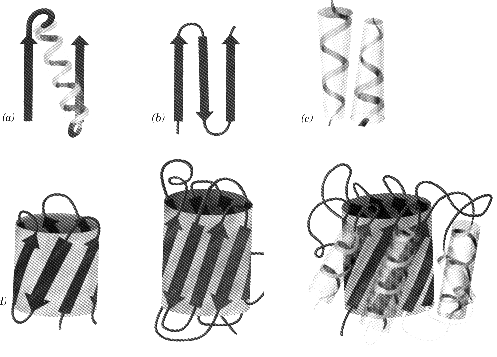
Motifs grouping of secondary structural elements
Common Motifs bab most common motif b-hairpin antiparallel strands connected by tight turns aa motif two successive antiparallel a-helices pack together
b-barrels collection of antiparallel sheets align to form a barrel appearance
Domains exist within proteins with greater than 200 amino acids fold into a globular cluster give the protein a multi-lobed appearance independent unit that provides the protein with a particular function - often have specific functions binding sites are often found between the domains
Interaction of 2° structural units to form a native, globular protein. - See the figure below for an all a-helix protein folding into its native globular form teritary structure
Summary of Tertiary Structure Bonding
Quaternary structure proteins with more than one subunit are termed oligomers which are constructed from a set of monomeric subunits Nomenclature Monomer single subunit Oligomer multiple subunits Protomer identical units within the oligomer Dimer two subunits Trimer three subunits Tetramer four subunits
Summary of Protein Structure
Protein Folding and Stability Stabilizing Forces
Hydrophobic Effect causes non-polar compounds to minimize their interaction with water this is the MAJOR driving force in folding a protein into its native structure the hydropathy index describes the tendency of an individual amino acid the greater the hydropathic value the greater the chance the amino acid will lie in the interior of the protein - (detergents and organic molecules disrupt these types of interactions) Electrostatic Interactions overall an ion pair contributes very little to the overall 3-D shape of a protein the ion pair tends to lock the two sidechains leaving less freedom to move thereby decreasing the entropy of the system, in addition, the ion pair prevents solvation of each R-group with water Hydrogen Bonds the second most important type of interactions within a protein the bond between the two atoms develops as each shares electrons unequally hydrogen bond donor and acceptors come together Chemical Cross-Links disulfide bonds exist between nearby cysteine R-groups tend to be a stabilizing force within proteins disulfide bonds inside the cell are rare as the intracellular environment is a reducing most proteins with disulfide bonds are those that are secreted into the oxidizing environment surrounding the cell these proteins often act as defense mechanisms (they tend to be toxic toward our cells) metal ions also tend to lock proteins into rigid conformations
Protein Denaturation/Renaturation proteins can either be in their native (functional) state or in a denatured (in active) state Protein Denaturants Heat pH Detergents Chaotropic agents guanidinium ion or urea
Organic solvents
RNAse A Denaturation/Renaturation Experiment
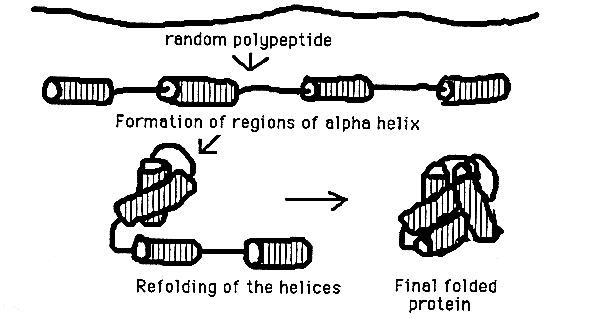
Protein Folding Pathway Step 1 secondary elements start to form Step 2 these elements start to collapse to bury hydrophobic amino acids - Molten Globule forms Step 3 the final 3-D shape of the monomer is formed Step 4 monomers associate with oligomers for the final protein structure
Secondary Structure Prediction (PELE)
History Pauling(1951) - postulated polypeptide chains could adopt alpha-helical or beta-sheet arrangements these predictions were based upon hydrogen bonding and cooperativity criteria Chou and Fasman (1974) designed the first simplistic algorithm for predicting secondary structural elements form primary sequence their technique made use of the 15 known protein structures at that time and some simples rules Rost and Sander (1993) trained a two-layered neural network on a non-redundant data base of 130 proteins to predict secondary structure utilized multiple sequence alignments as an integral part of their algorithm assumption that 3-dimensional structure diverges more slowly than primary sequence Wako and Blundell (1994) utilize multiple sequence alignments and substitution patterns between the various members of the group Burkhard (1996) utilizes profile based neural network to predict secondary structure again the basis is a reliable multiple sequence alignment
Techniques/Algorithms Chou/Fasman
Selected 15 known protein structures 2473 a.a. total break down where these 2473 a.a. were located (helix, sheet, coil) derive a normalization procedure to predict alpha common a.a. are glu, ala, leu, and his beta common a.a. are met, val, ile, cys coil common a.a. are gly, ser, pro, asn
Normalization Procedure frequency of certain a.a. within helix, sheet or coil = f = # a.a. in structure/total a.a. average frequency = <f> = summation of f for all a.a. in a category/20 a.a. - provides the frequeucy of each a.a. within either helix, sheet or coil
protein conformational parameter for each a.a.= P = f/<f> - provides a normalization factor for predicting whether an a.a. is found within helices, sheets or coils (P a= parameter to measure the propensity of a residue to be in the helical conformation) = values above 1.0 are indictors of strong preference to be within a certain secondary structural element (i.e. alanine has a P = 1.45) Other indicators of helix
N-terminal capping typically (Glu, Asp, Pro) C-terminal capping typically His, Lys, Gln, Arg Gly and Pro are rarely found within center of helix
Prediction using P a and Pb General Rule: need four helix formers out of six a.a. or 3 beta formers out of five a.a. are found clustered together in any native protein segment, the nucleation of these secondary structures begins and propagates in both directions until terminated by a sequence of tetrapeptides designated as breakers Assignment of 20 a.a.
Ha = strong helix former ha = helix former Ia = weak helix former ia = helix indifferent ba = helix breaker Ba = strong helix breaker (you can replace all of the a with b for the beta version)
Predictive Rules for Alpha Helix
any segment of 6 a.a. or longer in a native protein with Pa³ 1.03 and satisfying (1 4 above) is predicted as helix Predictive Rules for Beta Sheets
Rule 2: Problems
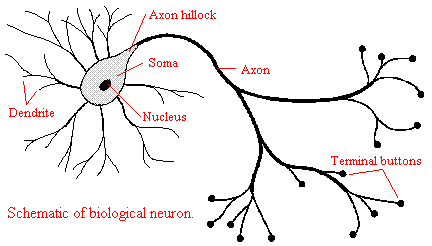
Neural Networks Biological Neural Network
Artificial Neural Networks - neural networks comprise a particular tool for pattern recognition researchers implement rules by providing knowledge to the network as a starting tool need strong training data- change the training data so less errors are made for protein structure prediction need high quality structural information and evolutionary information (multiple sequence alignments) one of the common features today is to note substitution patterns, which allow for identical secondary structure compare a multiple sequence alignment for a.a. substitutions compile these replacements and feed into neural network so the program can learn
Prediction of Motifs and Domains (PROSITE)
Definitions
Motif (Super-secondary Structure) organization of secondary structural elements into repetitive, regular elements such as helices and sheets all connected by intervening coils (loops) Domain A collection of motifs can be further arranged to provide a stable, discrete region of the protein responsible for a particular function
Programs for Motif/Domain Identification
PROSITE method of determining the function of an uncharacterized region of protein it is a database consisting of biologically significant patterns and profiles formulated in such a way as to identify to which family of protein the new sequence belongs, or which domain it may contain typically a cluster of residue types, which known to signify a motif of fingerprint can be identified - the use of protein sequence patterns and profiles to determine function will be the challenge for the genome project for years to come (Proteomics is born) Hydropathy Index (GREASE)
Definition
Index which assesses the hydrophilic and hydrophobic properties of each of the 20 a.a.
Programs for Hydropathy Index (GREASE)
the method utilizes a moving-segment approach that continually determines the average hydropathy within a segment the consecutive scores are plotted from amino to carboxy terminus
Details of GREASE
Choice of Hydropathy Index utilize experimental data for the free energy of transfer of a.a. from water to the ethanol in combination with the fraction of each a.a. found to be buried within x-ray crystal structures provides a numerical Hydropathy Index(Scale) see handout Table 2 individual Hydropathy Indexes were collected into cluters I, II, or III (the last digit in the index values seems to be of little importance)
Choice of Spanning Region 7 to 11 residue regions are spanned and the relative hydropathy indexes are added to provide an overall value for the region
Special Considerations for Membrane Spanning Helices additional constraints can be placed on the spanning region when searching for transmembrane helices facts we know (the typical bi-layer is 30 Å in width, if each turn of helix spans ~5.4 Å, then we need at least 5.5 complete turns of helix and if there are 3.6 a.a./turn then we need roughly 20 a.a to completely span the lipid bi-layer therefore the spanning region is increased to 19 a.a. when searching for transmembrane helices
Transmembrane Helix Predictions (TMHMM)
Definition prediction method which utilizes evolutionary information as input to neural network systems to predict secondary structure (including transmembrane helices) Details of TMHMM
Generate a Multiple Sequence Alignment crucial part of prediction process
Feed the Alignment into a Neural Network
|
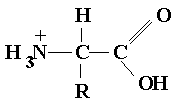


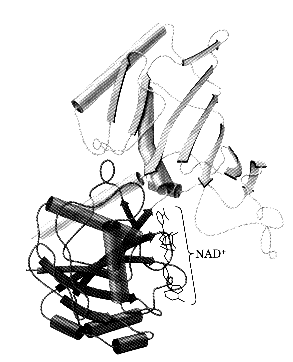
|
|
|