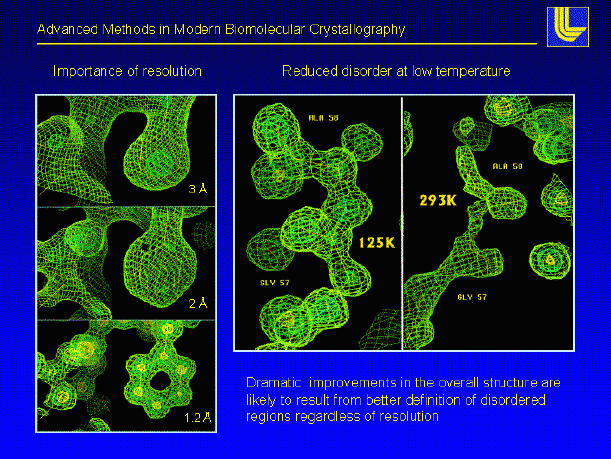
|
BIO 460: BioInformatics Protein Models
Methodology
Prepare protein sample
Crystallize protein sample
X-ray data collection
Build protein structure
Refine protein structure
Interpret 3-D model
Run Statistics
Deposit in Protein DataBank
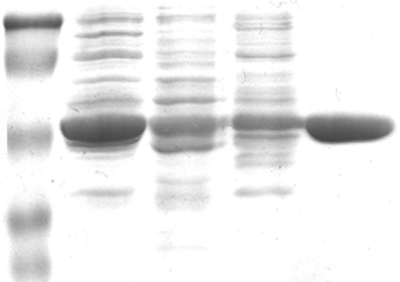
Protein Preparation the amount of protein needed for crystallize trials is significant; it is not uncommon for 1 mg of protein to be utilized per crystallization box (24-wells) in addition the protein sample needs to be purified to homogeneity
- Re-engineer protein for easy isolation and over-expression (pET vectors are commonly used)
- Design a purification scheme to provide homogenous protein sample
- Verify the purified protein/enzyme has retained biological activity
- Concentrate protein to ~10 to 200 mg/ml for crystallization trials
Protein Crystallization Trials
- Initiate crystal trials (companies have designed factorial screens)
- Screens are constructed with a variety of additives, which may induce protein crystallization
- Protein crystallization (fine line between nucleation/crystal formation
- You name the variable and it may effect protein crystal growth (pH, salt conc. Precipitant, protein conc., temperature, additives)
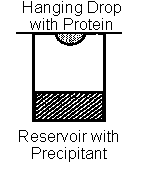
- Hanging drop vapor diffusion is a common technique to induce crystallization
Crystal morphology - crystals typically grow between 0.1 mm to over 1 mm in all dimensions unlike their inorganic counterparts, protein crystals are 30 70% water therefore these crystals tend to be very fragile
Hampton Research has a web page devoted to crystal growth
(http://www.hamptonresearch.com/stuff/gallery.html)
the interior of the crystal is constructed from repeating protein molecules the forces which hold a crystal together must overcome the natural tendency of disorder in the universe forces which hold a protein crystal together are noncovalent interactions between adjacent protein molecules
X-ray Diffraction
- X-ray crystallography is an experimental technique that exploits the fact that X-rays are diffracted by crystals. It is not an imaging technique. X-rays have the proper wavelength (in the Ångström range, ~10-8 cm) to be scattered by the electron cloud of an atom of comparable size. Based on the diffraction pattern obtained from
X-ray scattering off the periodic assembly of molecules or atoms in the crystal, the electron density can be reconstructed. Additional phase information must be extracted either from the diffraction data or from supplementing diffraction experiments to complete the reconstruction (the phase problem in crystallography). A model is then progressively built into the experimental electron density, refined against the data and the result is a quite accurate molecular structure.
The knowledge of accurate molecular structures is a prerequisite for rational drug design and for structure based functional studies to aid the development of effective therapeutic agents and drugs. Crystallography can reliably provide the answer to many structure related questions, from global folds to atomic details of bonding. In contrast to NMR, which is an indirect spectroscopic method, no size limitation exists for the molecule or complex to be studied. The price for the high accuracy of crystallographic structures is that a good crystal must be found, and that only limited information about the molecule's dynamic behavior is available from one single diffraction experiment.
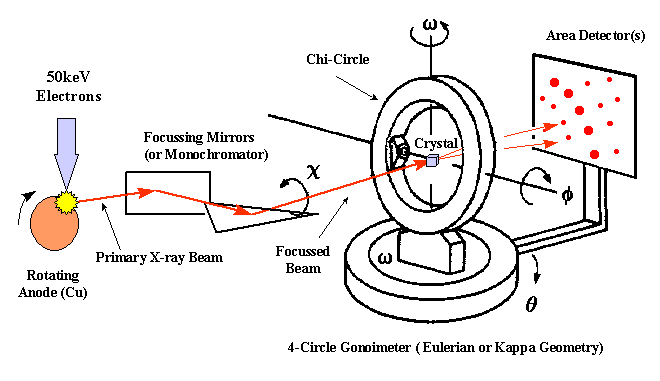
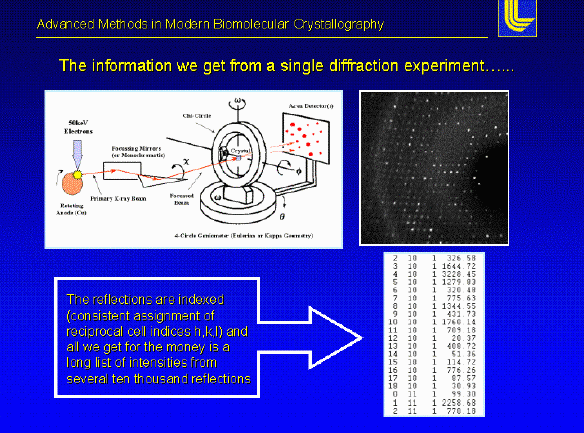
Experimental set-up - To perform a X-ray diffraction experiment, we need an x-ray source. In most cases a rotating anode generator producing a X-ray beam of a characteristic wavelength is used. The primary X-ray beam is monochromated by either crystal monochromators or focusing mirrors. After the beam passes through a helium flushed collimator it passes through the crystal mounted on a pin on a goniometer head. The head is mounted to a goniometer, which allows to position the crystal in different orientations in the beam. The diffracted X-rays are recoded using image plates, Multiwire detectors or CCD cameras.
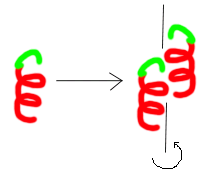
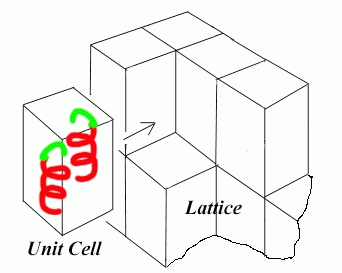
Arrangement of protein molecules within the crystal consider the motif illustrated below for a very simplified protein
- The motif can arrange itself via noncovalent interations to adopt numerous types of internal symmetry below I have an example of 2 samples of the same motif related by a two-fold screw
- Beyond the simple two fold screw the protein molecules will start to pack into higher order structures, which we can start to construct imaginary boxes around termed the unit cell if I know the unit cell and all symmetry relations relating each protein molecule to one another I can construct the entire crystal
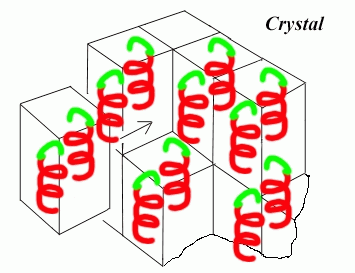
- The illustration below compiles the motif, symmetry, unit cell to construct the crystal we see under the microscope
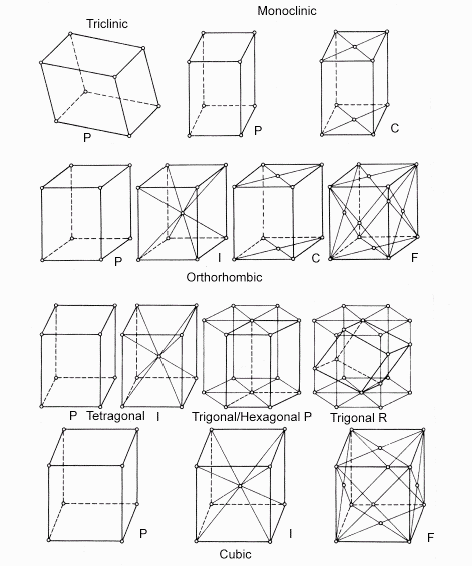
- The protein crystal built from the simple motif there are 7 crystal systems allowed for constructing the unit cell, which builds the final crystal (triclinic, monoclinic, orthorhombic, tetragonal, trigonal, hexagonal, and cubic)
- The data from an X-ray experiment is just a list of intensities, but to view the actual structure we need to know the phase for each diffracted wave. In other words the direction. To date there is no way to measure the direction of each diffracted reflection from a protein crystal. To solve this problem (termed the Phase Problem; the major obstacle in solving an protein structure), we have three basic techniques
- Multiple isomorphous replacement (soaking the crystal in a solution of heavy atoms (i.e. Mercury, Platinum, lead, uranium, gold, etc.) the metal will replace a light atom (typically a water molecule within the crystal). the heavy metal contain much more electron density than a typical light protein atom (carbon, nitrogen, oxygen, hydrogen) by collecting a set of data with and without heavy metal bound we can place the position of the metal within the protein crystal using the position of the bound heavy metal we can calculate the approximate phases for all reflections within the protein crystal
Molecular Replacement a technique which utilizes a known protein structure from a homologous protein to search for and replace into the unknown protein crystal the phases are derived from the rotated model once it has been fit into the unknown crystal
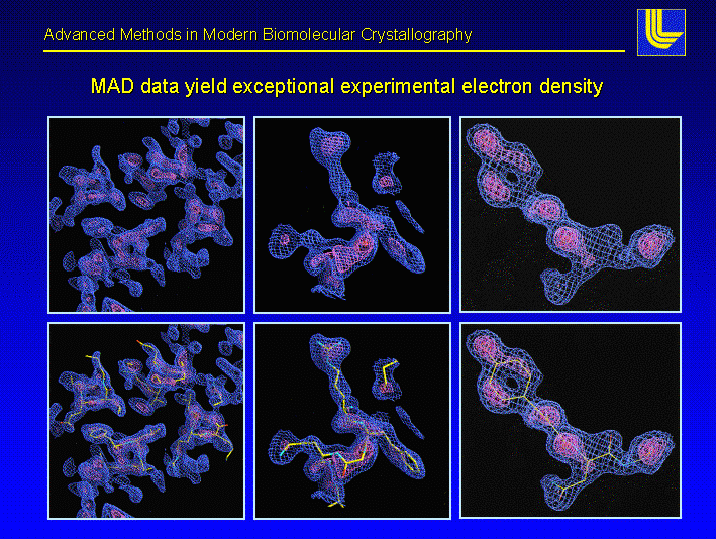
Multiple Anomalous Dispersion combines both molecular biology techniques and physics the protein of interest is expressed within the bacterial cell in minmal media supplied with 19 of the normal 20 amino acids methionine is replaced by selenyl-methionine (the sulfur is replaced by an atom of selenium) the selenium offers some unique properties it has unique absorption/emission properties at set wavelengths 4 different sets of x-ray data are collected a 4 different wavelengths on one crystal the beauty of this type of experiment is that the same crystal is used to solve the structure (it is said to be perfectly isomorphous) examples of the data we have after phases are collected to match the intensity data are illustrated below
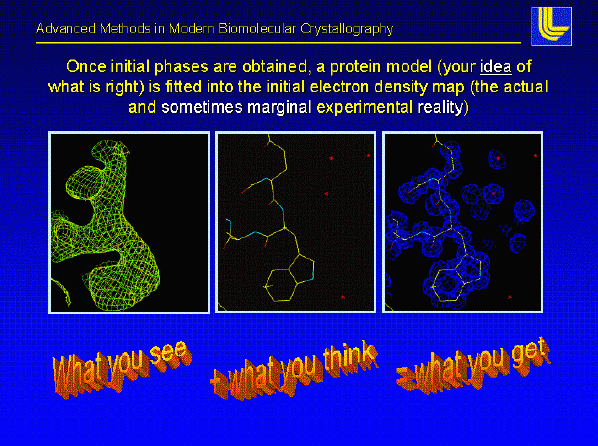
Once the phases are good enough the entire protein can be placed into the electron density map this is by far the most fun part of protein crystallography we utilize the resulting electron density and the primary sequence to build the protein model
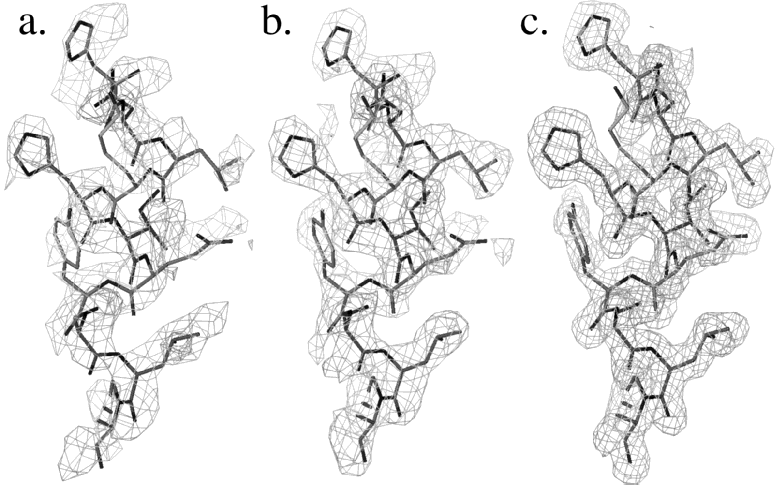
Once the model is constructed it will be placed into a computer program, which takes care of bond lengths and angles in addition, the program can place corrections on the bulk water surrounding the protein this technique is termed solvent flattening and it tends to enhance regions of poorly defined protein electron density iterations of refinement and model building are performed until the model lies is good agreement with the experimental data (the data we collect on the x-ray machine) XPLOR, CNS are common refinement programs in protein crystallography
In addition to protein atoms a crystallographer will add in well resolved water molecules (remember our protein crystals are 30 70 % water by nature) after adding the water molecules more refinement is performed new electron density maps are calculated and the protein model is further rebuilt the phases can then be calculated at the highest resolution your protein crystal diffracts the incoming X-ray beam
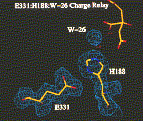
If an inhibitor can be added to the protein crystal and using the newly refined phases we can locate the position of the added ligand, and by default the location of the active site
Realize with static crystal structures this is only one "still" picture in the lifetime of a very dynamic biological molecule it may tell us a bit of the story, but not the entire story often times there are large conformational changes which occur within proteins inside the cell that are lost during the X-ray experiment still X-ray structures still provide the most information regarding the structure/function of protein
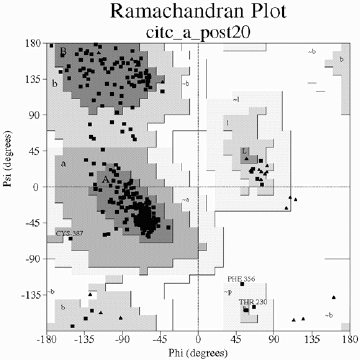
Once you trust your protein structure (meaning you have run your model through a series of programs which estimate the amount of error in the model (bond lengths, angles are assigned correctly, there are no large steric conflicts) you may deposit your protein model into the Protein DataBank for others to access it PROCHECK, WHATIF are common programs which search you built protein model and indicate errors based upon standard geometries the plot below represents all of the phi and psi angles for a final protein model we expect all r-groups to lie within the darkest shaded regions within the plot (either alpha-helix or beta-sheet) notice below we have some mis-behaving sidechains these are not always bad!
|