|
Current Level |
||||||||||||
|
|
||||||||||||
|
Previous Level |
||||||||||||
|
|
||||||||||||
|
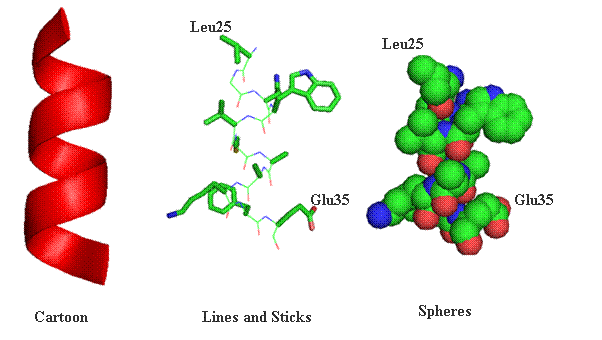
Lab 4.1 Proteomics The general outline for the last labs is to become familiar with the programs used to analyze protein sequences and structures using some protein examples. You will each be assigned a protein to explore individually, using the techniques we went through in lab. There should be time during lab for you to work on your individual projects, although some work may need to be done outside of class. This section focuses on proteomics, which is the study of protein structure and function using computers, databases, and protein sequences/structures. The first section provides a graphical review of protein structure from primary through quaternary levels. Protein Structure Protein structures can be represented a number of ways. Many times cartoon versions are used to emphasize where secondary structure elements exist. Below the same the segment spanning amino acids Leu25 through Glu35 have been represented in one of three ways (cartoon, lines and sticks or spheres). As a cartoon drawing, only the mainchain segments are represented and all sidechain information is left out of the final representation. This viewing option always for clarity and easily recognized secondary elements. Lines and stocks are often used for smaller section of a protein. The lines and sticks in a sense represent the bonds shared between atoms. In conjunction, each atom can be represented by a unique color. In the drawing below carbon atoms are green, nitrogen blue, oxygen red and sulfur yellow. Finally, protein structures can be represented where each atom is supplied as a sphere (volume). In this type of representation the compactness of protein can be witnessed.
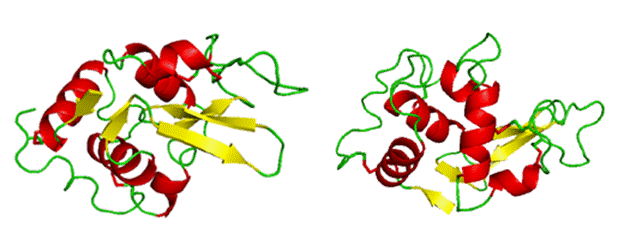
Lysozyme will be used to emphasize certain aspects of protein structure. Below is a cartoon version of lysozyme in two orientations. The cartoon drawing depicts various secondary structures colored by type where red (alpha-helices), yellow (beta-strands), and green (random coil).
Rhodopsin will be used to emphasize transmembrane domains at the end of this lab.
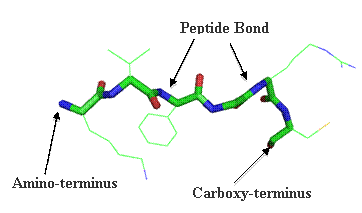
Primary structure (1°) is encompassed by the amino acid sequence. Adjacent amino acids are linked via a peptide bond. The NCBI web site has a useful amino acid analysis interfaces under the ALL Resources (A-Z) link and then under Amino Acid Explorer.
The primary sequence can be used to predict potential secondary structural elements. In addition, a number of primary sequences of the same protein from different sources can be aligned for identities and similarities. This type of alignment comparison is useful during the prediction of what are termed conserved domains. More on this later.
Primary Sequence Search There are many ways to search, import, view and analyze primary sequences from a variety of different databases. A very commonly used proteomics server is ExPASY. In the ExPASY site we could go under the databases section and find a protein sequence by name and organism. We can also search for a protein sequence using NDJINN within Biology Workbench. ExPASY Style
This web page provides a lot of information based upon the primary sequence you selected for lysozyme. The page provides name of the protein, length of sequence, organism derived, function of protein, important sites and amino acids within the protein and even a map for the secondary structural elements (more on this later). Biology Workbench Style
Once you have this sequence in your Protein Tools area you can subject it to a number of other programs within Biology Workbench.
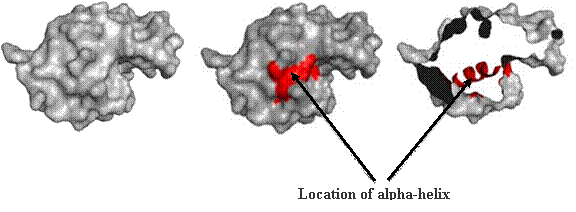
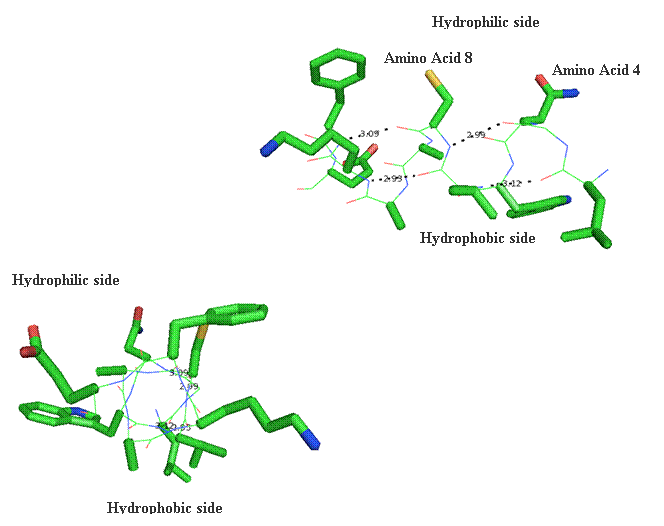
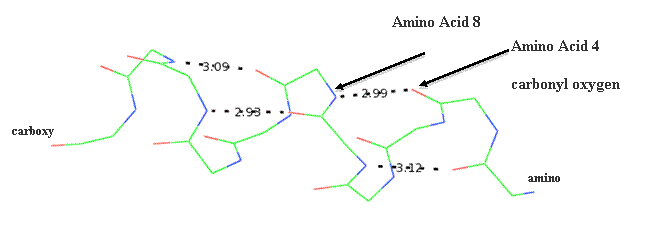
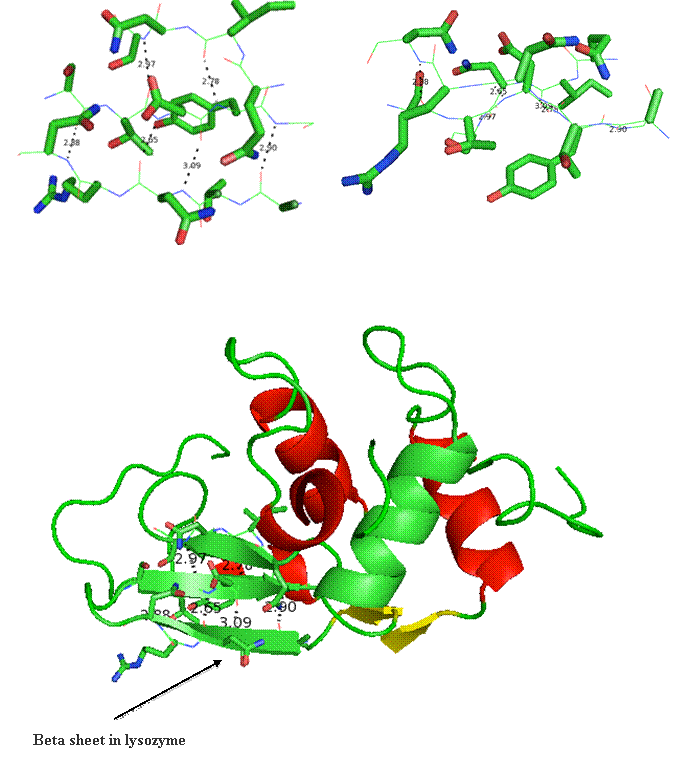
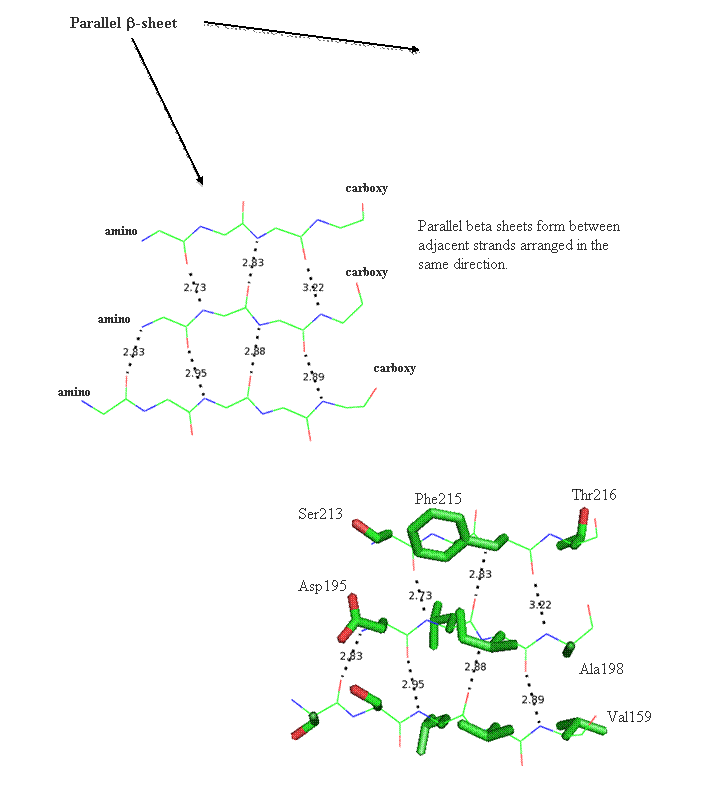
Secondary structure (2 °) forms via a repeating pattern of hydrogen bonds shared between mainchain NH and CO groups. There are two common forms of secondary structural elements, termed alpha helix and beta-sheet.
  
Alpha helices often have a sidedness to their appearance,
where one side is predominantly polar and the other is clearly hydrophobic. This
type of alpha-helix is termed amphipathic.
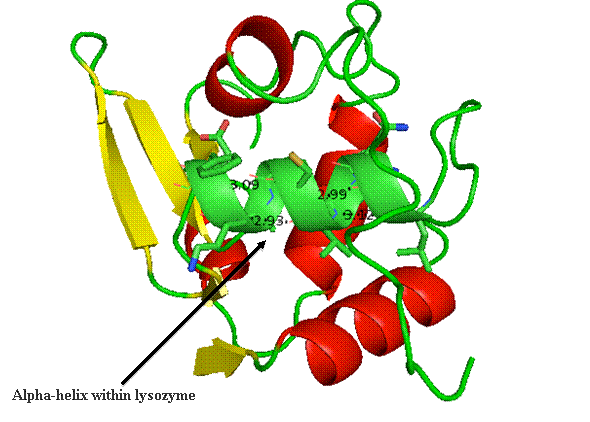
The alpha helix within lysozyme is shown as part of the
complete 3D structure. The under side of the helix harbors the hydrophobic face,
while the polar side projects out toward
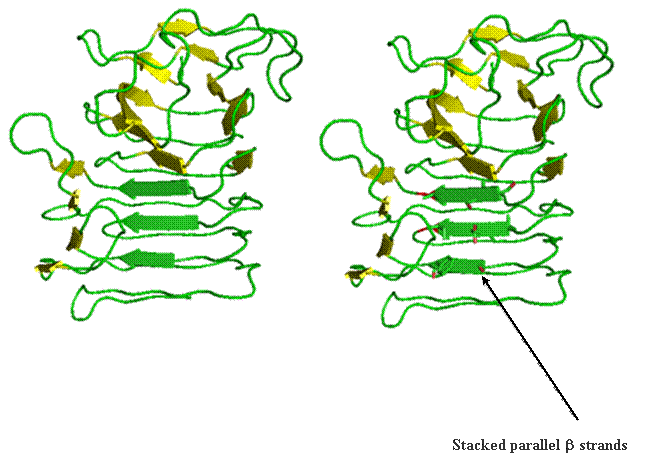
Beta helix structure is another way parallel beta-strands can be utilized to build tertiary and quaternary structure. The folding of this structure proceeds progressively from the top to the bottom by wrapping parallel beta-strands in coils. This is likened to wrapping wire around a pencil. The images below reports the coil form the parallel strands provide.
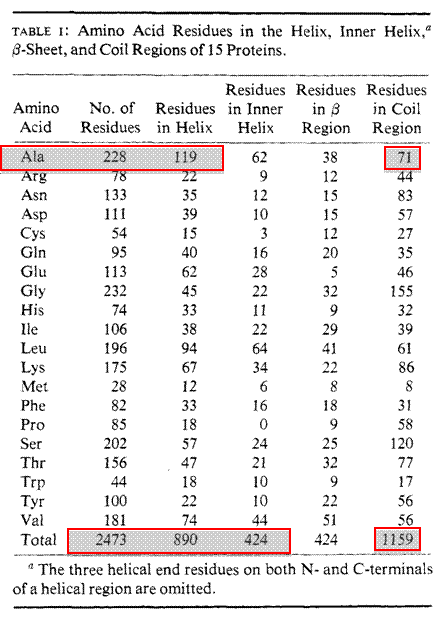
Secondary structure prediction can be made using a number of different programs. These programs have been written based upon many known 3D protein structures. Protein structures have been solved and deposited into a database known as the RCSB Protein DataBank (PDB) for over 33 years. The first 13 protein structures were solved and deposited in 1976 and as of January 1, 2010 over 62,000 protein structures have been determined and submitted to the PDB. One of the original algorithms written to predict secondary structure was designed by two scientisits Chou and Fasman. Their Chou-Fasman Method has been used successfully to predict many secondary elements from only the primary sequences.
Chou-Fasman Method
Experimental Dataset
Normalization Factor
Prediction Rules
Example Calculation
§ Frequency of alanine in helix, sheet or coil –228 total alanines (119 in helix, 38 in sheet, 71 in coil) –fa = 0.522 helix, fb = 0.167 sheet, fc = 0.311 coil
§ Average frequencies –<fa> helix = 890/2473 = 0.359 –<fb> sheet = 424/2473 = 0.171 –<fc > coil = 1159/2473 = 0.469
§ P value calculation –fa/<fa> = 0.522/0.359 = 1.45 for alanine (strong probability for helix) –fb /<fb > = 0.167/0.171 = 0.97 (lower probability for sheet) -fc /<fc > = 0.311/0.469 = 0.63 (low probability coil)
General Trends
Manual Secondary Structure Prediction § Assign a set of P-values to the following sequence(s)
Arg Asn Ala Glu His Lys His Ala Glu Leu Gly Pro Pa Pb Pc § Predict whether this span of amino acids is more likely to be alpha helix, beta sheet of coil
Computer Based Secondary Structure Prediction We can also use the EXPASY Proteomics Server and Biology Workbench to make predictions for secondary structure elements for an input primary sequence.
ExPASY Style
Biology Workbench Style
Tertiary Structure (3°) consists of collapse of the secondary elements driven by hydrophobic effect. The hydrophobic effect is explained by the placement of the non-polar amino acids into the interior of the finally folded protein. This increases the entropy of water and this is thought to be the driving force during protein folding. Bonding at this level involves sidechain or R-group interactions. Bonding
at this level includes the non-covalent bonding types: ion pairs, hydrogen
bonds, and hydrophobic interactions. In
Lactate dehydrogenase (LDH) is displayed in its monomeric and tetrameric forms as colored by secondary structure.
.
Patterns, Motifs and Domains Patterns or sites are small sections of consecutive amino acids that harbor a funtion or are a location subject to modification. Examples are phosphorylation (phosphate), glycosylation (sugar), and myristylation (fat) sites. These sites are redundant in proteins because they are defined by only a few amino acids. Thus, within a typical protein having ~200 amino acids the odds of finding a three amino acid sequence is common.
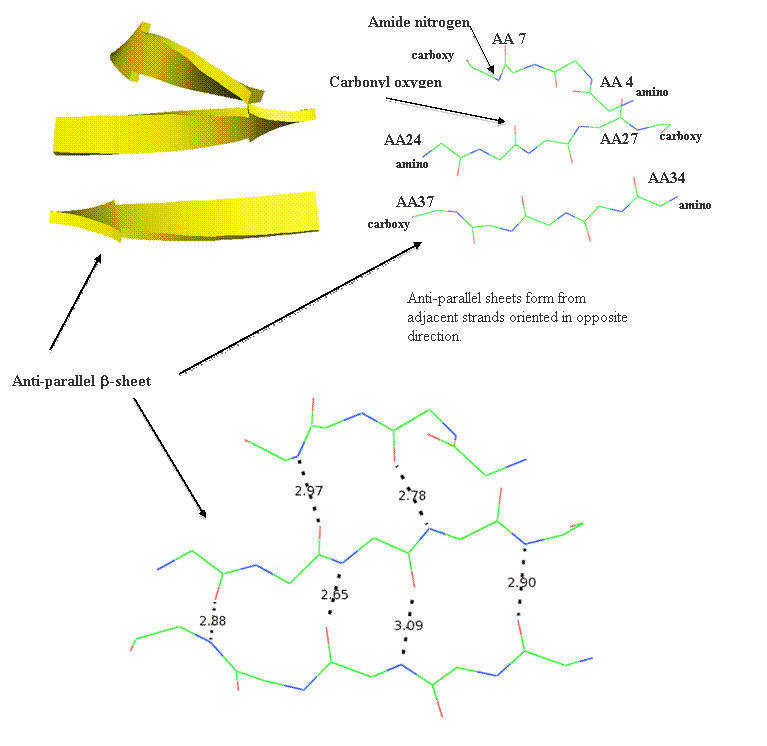
Motifs (or super-secondary structure)are built from simple arrangements of secondary elements and typically only structural. Some commonly recruited motifs are structural and include the beta-hairpin, the Greek Key, the Zinc Finger, the beta-alpha-beta motif, and the alpha helix-turn-alpha helix motif. These structural elements are stable and used to connect beta-strand elements. The beta-hairpin is used to connect adjacent anti-parallel strands, while the beta-alpha-beta motif connects parallel strands.
Some motifs are built from much smaller arrangements of amino acids that are quite far apart in the primary sequence. For example, the trypsin family catalytic triad contains a Aspartic acid-102 , Histidine-57, and Serine-195 located within the active in close proximity. However, from the priary structure one would not predict that these three residues lie within close proximity to form a functional motif within the trypsin active site. It was only after a number of other biochemical and structural studies were conducted that allowed for these three residues to be grouped into the trypsin family catalytic triad motif. A number of programs like PROSEARCH and PPSEARCH will search the PROSITE database for motifs and patterns.
Domains are known as functional units within proteins. Size ranges for domains span from a low of 36 amino acids up to 692 amino acids. The majority of domains have less than 200 amino acids and the average domain harbors 100 amino acids. Conserved domains are functional regions within proteins that pieced together during molecular evolution. In this fasion, new proteins with different sets of functions can be generated over time leading to evolutionary changes. These types of domains appear as clusters of amino acid sequence. In fact, these unique arrangements of amino acid clusters are usd to identify the so-called conserved domain. Multiplie sequence alignments provide information regarding where plausible conserved domains lie in a protein sequence. A second type of domain classification is 3D domains. These domains are based upon known and conserved three-dimensional shapes of proteins. 3D domains are recruited during evolutiona as stably folded and functional units. A conserved domain may not yet have a representative 3D domain. A 3D domain prediction requires a known 3D structure be proved during the comparison. The program SMART-Simple Modular Architecture Research Tool can be used to find functional domains in primary sequences.
In the upper corner of the RCSB homepage the PDB Stastistics link provides some interesting information regarding the various protein structures deposited to the RCSB PDB. One of the more interesting pieces of information is in the number of folds determined by year. As you can see by the mid-1980s there was a steady increase in the number of total protein folds. This increase reached a peak in 2007.
Pattern, Motif and Domain Prediction There are a number of programs available to search primary sequence for sites, motifs and domains. These programs sift through databases that have compiled sets of unique sites, motifs, and domains. These databases grew rapidly in the 1980s and 90s based upon functional and structural studies on many new proteins. Lysozyme ExPASY Style
Biology WorkBench Style
Entrez Style
SMART Style
Pyruvate Kinase Pattens and Motifs ExPASY Style
Domains SMART Style § Go to the SMART site § Submit the pyruvate kinase sequence for analysis under the PFAM domains
Some proteins have a portion(s) of their amino acid sequence embedded within the lipid bilayer. These areas of the protein sequence that are embedded within a bilayer must be hydrophobic. There are bioinformatic programs that are able to predict the hydrophobicity of an amino acid sequence. Again both ExPASY and Biology Workbench have some these programs accessible. BIOLOGY WORKBENCH contains three programs for determining regions of hydrophobicity in a protein and potential membrane spanning domains. Enter the Biology Workbench and select your sequence under Protein Tools. Next select one of the programs listed below. GREASE allows you to generate Kyte-Doolittle Hydropathy Profile. This does not predict secondary structure, so it will detect both alpha helix and beta sheet transmembrane domains. Numbers grater than 0 indicate increased hydrophobicity, numbers less than 0 indicate an increase in hydrophilic amino acids. TMHMM allows you to predict the location of transmembrane alpha helices and the location of intervening loop regions. This program will also predict which loops between the helices will be on the inside or outside of the cell or organelle. This program will not detect beta sheet transmembrane domains. It takes about 20 amino acids to span a lipid bilayer in an alpha helix. Programs can detect these transmembrane domains by looking for the presence of an alpha helix 20 amino acids long, which contain hydrophobic amino acids. TMAP uses a Kyte-Doolittle Hydropathy Profile to detect transmembrane spanning domains. This does not require that the domain be an alpha helix, as in TMHMM. It also provides the amino acid numbers for the transmembrane domain. This is especially useful for detecting signal peptides. A signal peptide is a short hydrophobic sequence at the amino terminus of eukaryotic proteins targeted for the endoplasmic reticulum and often for secretion.
Transmembrane Prediction A prediction of hydrophobic regions of proteins is based upon the Hydropathy Index. Numbers greater than zero indicted hydrophobic nature, while those values less than zero indicate hydrophilicity. Lysozyme § Under BIOLOGY WORKBENCH, go to protein tools § Select lysozyme § Run through GREASE, TMHMM, and TMAP Grease Output
TMAP Output PREDICTED TRANSMEMBRANE SEGMENTS TM 1: 6 - 28 (23)
TMHMM Output 0_LYG_CHIC Length: 211 0_LYG_CHIC Number of predicted TMHs: 1 0_LYG_CHIC Exp number of AAs in TMHs: 20.46703 0_LYG_CHIC Exp number, first 60 AAs: 20.18892 0_LYG_CHIC Total prob of N-in: 0.45484 0_LYG_CHIC POSSIBLE N-term signal sequence 0_LYG_CHIC TMHMM2.0 outside 1 9 0_LYG_CHIC TMHMM2.0 TMhelix 10 32 0_LYG_CHIC TMHMM2.0 inside 33 211
Rhodopsin § Under BIOLOGY WORKBENCH, go to protein tools § Use NDJINN to locate files containing the protein sequences of rhodopsin (HSU49742) § Use the GBPRI to only search primate sequences § Import this sequence and run through GREASE, TMHMM, and TMAP
PREDICTED TRANSMEMBRANE SEGMENTS TM 1: 45 - 71 (27) TM 2: 75 - 99 (25) TM 3: 114 - 142 (29) TM 4: 150 - 178 (29) TM 5: 203 - 231 (29) TM 6: 257 - 277 (21)
TMHMM Output
Report for Unit 4 (50 points total) We would like a formal written report with the following information. Don't paste in the questions, these are just to help you be organized. You can create figures in your report by right clicking on an image, and then copy and paste it into your report. Don't add lots of extra output, i.e. names and accession numbers from Biology Workbench, just the figure and a figure legend, and then explain what it means in your well-written, rational report. 1. Perform a BLASTP on your assigned sequence against the PDBFINDER (sequence of the protein from a crystal structure) and SWISSPROT-HUMAN (sequence from the DNA) databases in Biology Workbench (you can select both simultaneously using the Ctrl key) or use BLASTP at the ExPASY site directly.
2. A PyMol image containing the 3D structure of your protein in cartoon in three forms.
3. A description of the family of proteins to which your protein belongs (paralogs, Lab 4.2). For this portion of the unit we would like you to identify and import 6-7 related human protein sequences (not 6-7 different sequences of the same protein) and align these sequences. Try to choose several paralogs, not just the most closely related, but don't go much below score of 100 or the alignments won't be very good. If there is a known motif for the class of protein your protein falls into, look for this motif in your aligned sequences.
4. A description of the evolution of your protein in different species (orthologs, Lab 4.2). For this portion of the unit we would like you to identify and import 6-7 protein sequences of this same protein from different species and align these sequences. Try to choose some distantly related species for comparison, i.e. can you find this protein in yeast or bacteria? If there is a known motif for the class of protein your protein falls into, look for this motif in your aligned sequences.
5. A conclusion summarizing your findings. Specifically comment on the following Where is the most sequence similarity seen on the 3D structural alignments? Were orthologs or paralogs more highly conserved? Is this consistent with the relative functions of orthologs and paralogs?
Note: When writing this report do not simply attached the output from various programs at the end. You MUST embed all output within the report and near where you are discussing its relevance. This will take some organizational work on your part. It makes no sense to talk about the patterns, motifs, and domains on page 1 of the report and have the supporting output on page 5. I will NOT accept reports written where the images from various programs are simply added on to the end.
|
|





















|
|
|