|
Current Level |
||||||||||||
|
|
||||||||||||
|
Previous Level |
||||||||||||
|
|
||||||||||||
|
Molecular Modeling of Protein Families Now that we have identified the sequences of several members of the family of serine proteases, we will examine how these sequence differences influence protein structure. We have already aligned these sequences and examined gaps or loops present in some. We have also seen that the residues near the active site are conserved. We can get a better picture of this by examining the three dimensional structures of these proteins. To do this we will align all of the sequences with the amino acid sequence of a protein that has been crystalized. We can then view this alignment in 3D, as it would appear in the actual protein. We have seen how we can use computers to analyze the primary sequence of a protein to try and predict some of its properties (activity, sites of secondary modification, secondary structure, hydropathy profiles, etc.) In addition to these properties, we would also like to view our newly discovered protein in 3D. This requires either solving the structure of the protein by X-ray crystallography or NMR, both time consuming procedures which require a lot of protein. Alternatively, if we know that the 3D structure of a related protein has already been solved, then we can use molecular modeling to approximate the 3D structure of our new protein.
There are four steps to this process. 1. Find a related protein for which the 3D structure is known 2. View the structure and identify important domains such as the active site. 3. Align the primary sequences of the proteins with the sequence of the protein of known structure 4. Superimpose the amino acid sequence of the new protein onto the 3D structure of the known protein.
1. Obtain a structure of one protein in the family First we need to locate this structure. These are saved in several locations, we will use the Brookhaven Protein Database (PDB). If you already searched your assigned sequence against the PDBFINDER database you already have the structural accession number. Protein structures are also given identification codes. Record the code for your structure. Once you enter Protein Explorer you will use this to upload the structural file. For example, the accession number for trypsin is 1TRY. If you don´t know the acession number, or the name of the protein, you can perform a BLASTP search for the protein against the PDBFINDER database. If there is a structure accompanying the protein sequence it will be linked to the right of the sequence as Structure. 1. Go to the Biology Workbench (BW). 2. Select the session containing your protein sequences. Select "Resume Session", and press the [Run] button. 3. Press the [Protein Tools] button. 4. Select your assigned sequence and "BLASTP", and press the [Run] button. 5. Check "PDBFINDER". 6. A list of sequences is displayed. Each is prefixed with "PDBFINDER" (meaning it has a published 3D structure) pick a few sequences without spending too much time and effort on the selection process. Use the [Show Records] button to get more information about the checked sequences. 7. Import the Sequence 1TRN.
2. Viewing structures.
3. Align the sequences Before we can superimpose the sequences onto the 3D structure of one of the proteins we need to align these 6-7 sequences. One of the sequences in the alignment must be the sequence of the amino acids in the 3D structure. 1. Open BIOLOGY WORKBENCH and check any sequences you want to include in the alignment, including the trypsin sequence we just imported. This ensures that we have at least one sequence for which a 3D structure is available. Any PDBFINDER sequence has a 3D structure. 2. Run CLUSTALW 3. Examine the alignment carefully. An alignment that has very few identities, or very few differences, may not be informative. If you wish to exclude one or more sequences, press the [Return] button and rerun the alignment. 4. Once you are satisfied with the alignment, press [Import Alignment]. 5. You should now see a list of all alignments you have made, each with a checkbox. Notice that you are now in the Alignment Tools, no longer in Protein Tools. 6. Now we need to get the alignment in FASTA format. Check the checkbox for the desired alignment. Select "View", press [Run]. 7. At the top of the page, change the format to "Fasta". 8. Select and copy the alignment (hold down the left button on the mouse and scroll down the aligned seqeuence). 9. Paste into a word processor and save it as a text (.txt) file for later use.
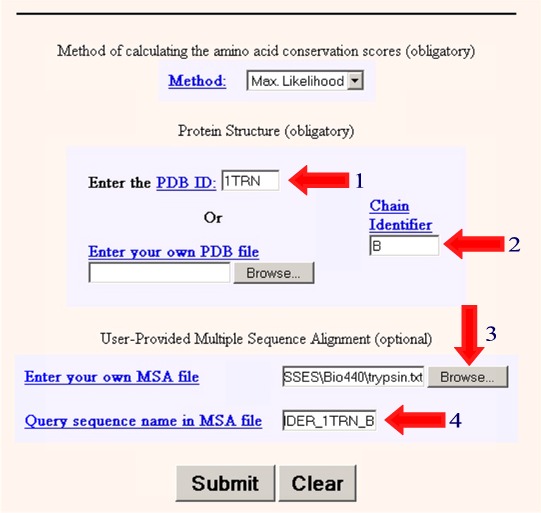
4. Superimpose the amino acid sequence of the new protein onto the 3D structure of the known protein Next we wish to take our aligned sequences and superimpose them onto the 3D structure. We will do this using the program ConSurf. We need to enter the following information information into the program. 1. Enter the PDB ID: 1TRN 2. Enter the Chain Identifier: B 3. Browse and find the text file containing your multiple sequence alignment. (eg. trypsin.txt) 4. Enter the name of the sequence in your multiple sequence alignment that matches the PDB crystal structure (PDBFINDER_1TRN_B) in this case.
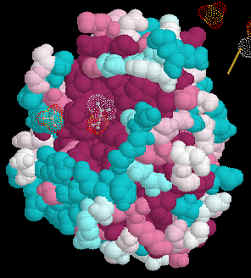
5. Select Submit 6. Wait a few minutes to get your results in Protein Explorer. 7. When you have the image as you would like it select Edit/Copy to copy the image and then paste it into your report. If that doesn't work, try Ctrl/Print Screen to copy the entire monitor screen. Examine the 3D Alignment
Amino acids which are identical in all of the aligned structures will appear dark
purple. Those which are similar in light blue.
We aligned 6-7 amino acid sequences of related human proteins from one family. We then superimposed these aligned sequences onto the structure of one of the members of the family. This allowed us to study conserved regions of the protein in 3-dimensions. We can use the same tactics and techniques to study the evolution of proteins in different species. To do this we would select the same protein in several species, align the amino acid sequences and superimpose them onto the 3D structure of the protein from one species. An easy way to obtain sequences of the same protein from different species is to either use Biology Workbench and select specific databases to search, i.e. one each from mammals, invertebrates, plants/fungi and bacteria. Another method is to perform an Entrez search and then use Blink to identify related sequences. The program colors sequences from different kingdoms in different colors making it easy to identify distantly related species.
Your Assignment Search PDBFINDER using BLASTP for a 3D structure of the protein you were assigned.. Be sure to import this amino acid sequence so that you can align this sequence with the other sequences. Obtain 6-7 amino acid sequences of human proteins from the same family as the protein you were assigned. Obtain 6-7 amino acid sequences of the protein you were assigned from different species. With each group of proteins, perform a Clustal W alignment as before and use Consurf to create a consensus structure of this protein. We would like at least three images printed out from this exercise. Once you have generated your model you can either right click on the image and paste it into your final report, or use "print window" to make a hard copy of the molecule. Unfortunately the Consurf models cannot be saved. The PyMol models can be saved. These images will be part of your take-home assignment. Image 1. The PyMol image of your protein in cartoon form with the secondary structures colored and the active site residues in space filling format. You can probably identify the active site residues using PROSEARCH. Image 2. The Consurf image of your protein with the aligned sequences of the other family members superimposed. You will make a similar image of the aligned sequences of your protein from other species to illustrate evolutionary changes. Image 3. The Consurf image of your protein with the aligned sequences of the other species superimposed. You will make a similar image of the aligned sequences of your protein from other species to illustrate evolutionary changes.
|
|
|
|
|