|
Current Level |
||||||||||||||||||
|
|
||||||||||||||||||
|
Previous Level |
||||||||||||||||||
|
|
||||||||||||||||||
|
There are many structural elements (motifs) that are conserved among different proteins. For example carbohydrates can be attached to the amino acid asparagine in proteins through N-glycosylation sites which are indicated by the consensus sequence Asn-Xaa-Ser/Thr. The first amino acid is Asparagine (Asn), the second amino acid can be any of the 20 amino acids (Xaa), and the third amino acid is either Serine (Ser) or Threonine (Thr). However, just because this consensus sequence appears does not mean that the site is glycosylated. You can also look for more complex motifs or domains, such as enzyme active sites and receptor binding sites. We will look at four different programs.
The following three can be accessed through BIOLOGY WORKBENCH.
CDART: Conserved Domain Architecture Retrieval Tool. This program gives an interactive graphical display of the conserved motifs found in an amino acid sequence. You can click on each domain to learn more about its properties and consensus sequence. The program also provides graphical displays of all known proteins containing at least one of the domains found in your protein. One drawback is that this program only reports major domains, and not smaller motifs, and has fairly brief descriptions. It is a good place to start, but the programs described below under BIOLOGY WORKBENCH are more descriptive and thorough.
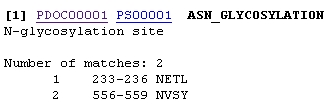
1. The program PROSITE analyzes a protein sequence for these known motifs and gives a description of each. This is useful when analyzing the sequence of a new protein to try to gain clues to its function. http://www.expasy.ch/tools/scnpsit1.html or through PROSEARCH in BIOLOGY WORKBENCH Enter the amino acid sequence that you wish to analyze or the accession number of the protein and press Start the Scan. You will be given an output which lists several motifs present in the protein, indicating the sequence that was identified and its position in the protein. Each will also contain a link to more information on that particular motif. For example the sequence being analyzed has potential N-glycosylation sites at amino acids 233 and 556. By clicking on PDOC00001 more information on N-glycosylation will be provided.
Other motifs are more complex and can include sites that bind cofactors or substrates (active site). Such information would be valuable in identifying the function of a protein.
2. RPSBLAST performs a blast search of your sequence vs. a database of conserved domains in families of proteins. Your sequence is compared to the consensus sequence of many families of proteins to look for a match. This is very useful in identifying which family your protein belongs to, especially over larger domains. For example, if we sumbitted a serine protease we would get the following matches.
If we click on the link smart00020 we would learn about the consensus sequenced used, information on the family of proteins, and other sequences which are closely aligned to our sequence. There is a new 3D imaging program which allows one to view the aligned sequences. This is not loaded on our computer, but we can view it as an html image.
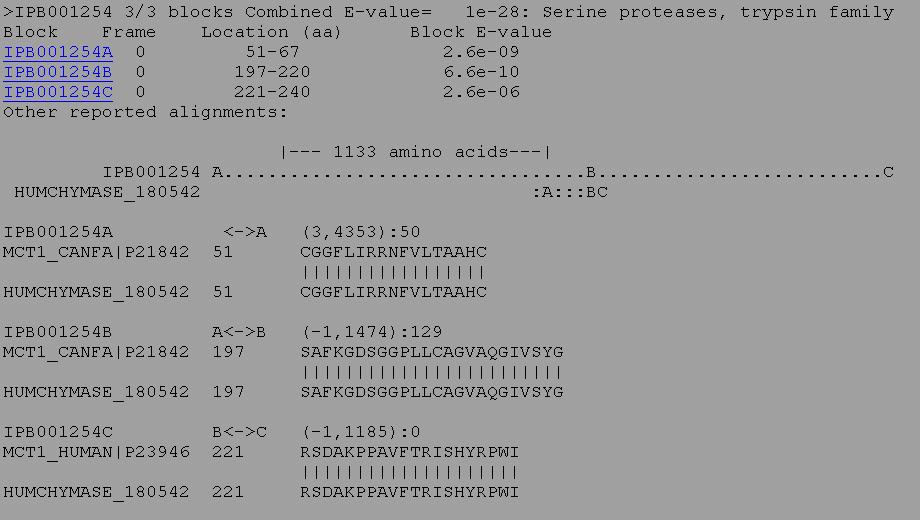
3. BLIMPS is similar to RPSBLAST, except that it looks for specific blocks or domains of sequence similarity. A protein may overall have relatively low similarity to another protein, but if it has high similarity in specific important regions it may have the same activity and be a homologous protein. BLIMPS compares a protein or nucleic acid sequence against an the BLOCKS database of conserved protein motifs. The scores for high scoring BLOCKS found within the query sequence are totalled and a family classification is made based on the total score for each block found in the query sequence. Individual block scores are listed beneath the family classification along with the highest scoring alignments. For example, the protein below matched 3 out of 3 blocks for the conserved sequence of an active site of a serine protease.
|


|
|
|