|
Current Level |
||||||||||||||||||
|
|
||||||||||||||||||
|
Previous Level |
||||||||||||||||||
|
|
||||||||||||||||||
|
Background on Genomics The study of entire genomes is a relatively new field which promises to unlock many secrets in biology. These include the identification of disease causing genes, generation of more accurate evolutionary trees and understanding how cell responds to changes in its environment at a molecular level. All of this work is based upon our ability to align and sequence very large pieces of DNA to create a complete map of each chromosome found in an organism. To generate a complete map, first individual overlapping clones are arranged to produce contiguous regions of a chromosome (called a contig). By overlapping many clones of 50-250,000 bases, we can generate contigs that span millions of bases on a chromosome. Because these clones contain so much genomic DNA they cannot be carried on traditional plasmids, but instead are cloned into special artificial chromosomes. These can include Bacterial Artificial Chromosomes (BAC), Yeast Artificial Chromosomes (YAC), and P1 phage Artificial Chromosomes (PAC).
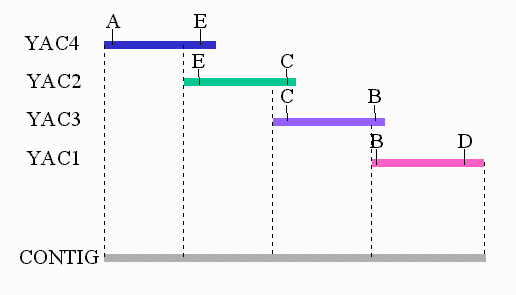
To determine whether two clones overlap we examine each clone for the presence of sequences of DNA that are unique throughout the genome. These sequences are known as sequence tagged sites ( STS) and "expressed sequence tag" (EST) sequences. An STS or EST is defined by two short synthetic sequences (typically 20 to 25 bases each) that have been designed from a region of sequence that appears as a single-copy in the human genome. These sequences can act as primers in a polymerase chain reaction (PCR) assay to score for presence or absence of the site in any DNA sample. In the example below, YAC4 contains STS ``A" and STS ``E", because a PCR product is formed when YAC4 is assayed with primers for STS ``A" and STS ``E".
If you assayed each of the four YACs in the example above with primers for each of the five STSs, you would generate the table below. This information would allow you to construct the map and contig shown above. A ``+" indicates that a PCR product would be formed using the indicated primers and YAC.
Sources of Sequences STSs can be designed from nearly any small fragment of DNA, but only about 3% of the bases in the genome actually correspond to genes. Transcribed sequences (i.e. mRNAs) are the only readily available source of this 3% of the genome. Full-length mRNA sequences for approximately 5000 human genes may be found in GenBank and partial "expressed sequence tag" (EST) sequences are available for tens of thousands more. There are many genome projects in progress, and the complete sequences of several simpler organisms are completely known. Each of these will have a unique set of STS and EST markers.Electronic PCR Because STSs are defined by sequence, it is possible to find these mapped landmarks in DNA sequences using a computational procedure known as "electronic PCR" (e-PCR). This provides a simple means to verify associations between STSs and the original sequences from which they were derived. More importantly, mapped sites may be found in other cDNA sequences for the same gene or in the rapidly accumulating human genomic sequence data. Old version (allows you to upload files and search for markers in other genome projects) New version (uses current names for STSs and has links to human genome project)
Four segments of human genomic DNA are linked below. Use the electronic PCR programs above to identify the STSs present on each segment of DNA. Use these STSs to draw a map of each segment of DNA, and combine any overlapping segments into a contig using a word processor as a sequence editor. Finally, use the intron/exon splice site identification program to look for the presence of any genes in this contig. As a follow-up exercise you could perform the same analysis on other fragments of genomic DNA from humans or other species. |

|
|
|
|||||||||||||||||||||||||