|
Current Level |
||||||||||||||||||
|
|
||||||||||||||||||
|
Previous Level |
||||||||||||||||||
|
|
||||||||||||||||||
|
An alignment program is used to compare the sequence homology between two protein or DNA sequences. These programs find the best match between the two sequences. Occasionally gaps need to be introduced to make the two sequences align. seq1 > 1 ggcctctgcctaatcacacagat-ctaacaggattatttc This type of analysis is useful in detecting evolutionary differences between species and to look for mutations in genes. We can either use the program BLAST directly to align two sequences, a Multiple Alignment Program or use the Biology WorkBench
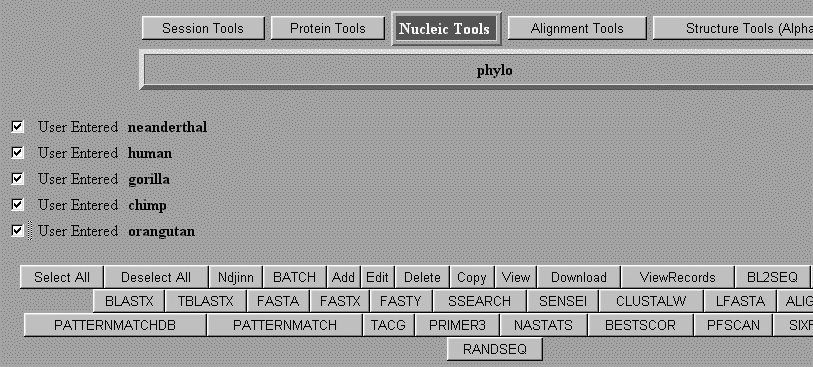
Using Biology WorkBench to align two or more sequences. Log onto the Biology WorkBench and either create a new session or resume an existing session. Select Nucleic Tools or Protein Tools. Select the sequences that you wish to align and then scroll down to CLUSTALW.
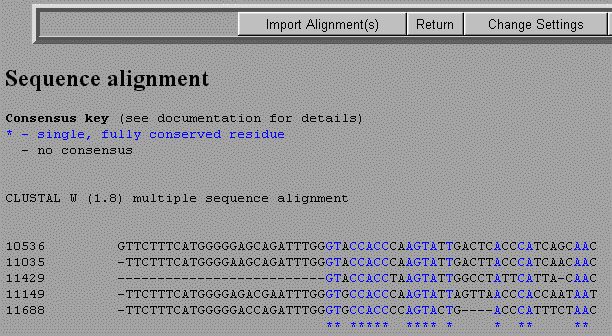
You will now be given some options on parameters you can change in your alignment. You can just use the default values and select Submit at the bottom of the page. The results will show the two sequences with colored letters representing a consensus. Black letters will illustrate a mismatch and dashes will represent gaps.
If you wish to use this alignment to create a phylogenetic tree, or just want to save the alignment, select Import Alignment(s).
1. The following program allows you to align two sequences together. RESULTS If you align DNA sequences, vertical lines will indicate identical bases and "-" will indicate gaps in the alignment. 1. To align several sequences the following Multiple SequenceAlignment program is useful. All of the sequences are entered in the same data box, with titles separating each sequence.
RESULTS The results from the multiple alignment will be given in two formats. The second is a FASTA format that can be pasted into some evolutionary programs. A " - " indicates a gap in the alignment of the two sequences. |
|
|
|